
Sometimes, when something is broken, the first step to fixing it is to break it even more.
In a recent example, scientists seeking to understand the mechanism of a DNA-repairing protein have discovered that the molecule performs its functions by first marking and then further breaking damaged DNA. The team’s surprising findings on the protein, called XPG, have provided much-needed insight into how DNA repair works in healthy human cells, as well as how different mutations can translate into different diseases and cancer.
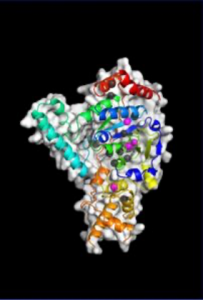
“We saw that XPG makes a beeline for discontinuous DNA – places where the hydrogen bonds between bases on each strand of the helix have been disrupted – and then it very dramatically bends the strand at that exact location, breaking the interface that connects bases stacked on top of each other,” said Susan Tsutakawa, PhD, a structural biologist in the Biosciences Area at Lawrence Berkeley National Laboratory and first author on the work published in PNAS. “The bending activity adds to an already impressive arsenal, as XPG was first identified as a DNA chopping enzyme, responsible for cutting out nucleotide bases with chemical and UV radiation damage.”
Yet despite this knack for destruction, the team notes that XPG is more like a master sculptor than a demolition crew.
“An unexpected finding from our imaging data is that the flexible parts of the protein – which were previously impossible to examine – have the ability to recognize perturbations associated with many different types of DNA damage,” said co-author Priscilla Cooper, PhD, a biochemist senior scientist in the Biosciences Area at Berkeley Lab. “XPG then uses its sculpting properties to bend the DNA in order to recruit and load into place the proteins that can fix that type of damage.”
A protein with many jobs
Although the extent of what XPG does in human cells is still only partially understood, scientists have long known that the protein is essential to human health by observing the devastating symptoms that occur when it is missing or not functioning normally. Cockayne syndrome, a disease characterized by a progressive and ultimately fatal neurological decline that begins in infancy, and xeroderma pigmentosum, a condition of varying severity characterized by extreme sun sensitivity and greatly elevated risk of skin cancer, are both known to be caused by mutations in the gene that encodes XPG.
Fascinated by its many roles, Tsutakawa, Cooper, and John Tainer, PhD, the director of structural biology at the University of Texas MD Anderson Cancer Center and visiting faculty in the Biosciences Area at Berkeley Lab, have been collaborating on studies of XPG for 20 years. The trio, and their many colleagues, pool their expertise in structural biology, molecular imaging, biochemistry, and cell biology so that they can map the protein’s structure and interpret how its three-dimensional form interacts with DNA and other proteins. They had previously discovered that XPG often binds to damaged DNA without engaging its DNA cutting activity, but could not examine the protein in great enough detail to find out what it actually does in these instances.
After many years spent developing technology that could catch up with their ambitions, the team was finally able to build a precise model of XPG’s catalytic core – the region responsible for the DNA cutting activity – and produce images of the large, multiple-unit molecule’s overall structure using a trifecta of cutting-edge imaging technology.
They performed X-ray crystallography at Stanford Synchrotron Radiation Laboratory, and small angle X-ray scattering (SAXS) at the SIBYLS beamline of Berkeley Lab’s Advanced Light Source. SAXS is a technique that has recently evolved to allow scientists to analyze flexible molecules moving freely between their natural states rather than in static or frozen conformations, as necessitated by crystallography. Such an approach is sorely needed for a protein like XPG, whose catalytic core is only one-quarter of the total structure and the rest is made of highly flexible “disordered” regions with no default shape.

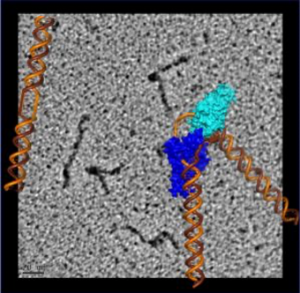
To visualize the XPG-bound DNA, the scientists recruited Jack Griffith, PhD, a pioneer of rotary shadowing electron microscopy at UNC Lineberger Comprehensive Cancer Center. Rotary shadowing electron microscopy allows direct visualization of individual DNA molecules with proteins bound to them, including how they were bent by XPG.
“The ability to see the shapes of individual DNA molecules gave us an essential clue as to how XPG works to identify and process damaged DNA,” said Griffith, a professor of biochemistry and biophysics and expert in protein-DNA interactions.
The electron microscopy imaging also provided visual evidence supporting the scientists’ previous surprising finding that XPG plays a role in homologous recombination – a DNA repair process frequently used by cells to fix dangerous double-strand breaks before replication. This means that XPG could interact with known homologous recombination proteins such as BRCA1 and BRCA2, defects in which are known to cause cancer.
Meanwhile, crystallography performed on the catalytic core shed light on how inherited patient mutations in the gene for XPG can translate into severe protein dysfunction and different diseases. The team made and tested catalytic core proteins having each of the 15 known point mutations that cause either xeroderma pigmentosum or Cockayne syndrome, and found that these single amino acid substitutions can destabilize the entire protein, but to different extents. The properties of the residual mutant protein will determine which disease results. “This structure helps us understand the distinction between the two diseases,” said Cooper, “and it reinforces how complex the protein is.”
Invigorated by the new information, the team has already begun a study looking at XPG’s role in different cancers, as well as a follow-up structural study of the protein’s disordered regions to learn more about its DNA sculpting properties.
“The superb technical and collaborative strengths of Berkeley Lab and our partners made this multidisciplinary breakthrough feasible,” noted Tainer.
“But we would also like to highlight the contribution of patients and patients’ families,” added Tsutakawa. “So much of what we have discovered was made possible by them choosing to share their DNA sequences with the scientific community.”
The Advanced Light Source (ALS) and Stanford Synchrotron Radiation Laboratory are Department of Energy (DOE) Office of Science user facilities. The SIBYLS beamline 12.3.1 at the ALS is also funded by the NCI Structural Biology of DNA Repair Program and the NIH through ALS-ENABLE.
—Aliyah Kovner, Berkeley Lab