Initial Protocol Development
To proceed with successful clinical development, you need:
- Preclinical data and/or previous human experience
- Drug
- Secured from a pharmaceutical partner,
- Purchased commercially, OR
- Validated UNC manufacturing
Clinical Protocol Templates
Once you are ready to proceed with opening your study or writing a protocol to secure funding for your study, there are many steps involved in initial protocol development and activation of your clinical trial. The pages in this section take you through those steps:
- Writing Your Protocol
- Writing Your ICF
- Submission to the Funding Source (if applicable)
- Protocol Review Meetings (including Correlative Review Meeting)
- Protocol Review Committee (PRC)
- FDA – Drugs/Biologics
- FDA – Devices
- Activation of the UNC Site
- Activation of Multicenter Sites (if applicable)
Click to expand image

Special Considerations
Developing a diagnostic (even if the main focus of the protocol is to study a drug/biologic)?Remember that you will need to consider both drug and device regulations. You also need to consider the use of honest brokers to separate the clinical team and the correlative team.
Honest Broker: An individual or system acting on behalf of the researcher to collect and provide de-identified information/samples to the research team.
Make sure in your clinical protocol that you are clear that you are developing a diagnostic and how you plan to use that diagnostic:
- Future implementation in follow-up studies?
- Retrospective analysis?
- To return results to patients?
- To determine eligibility?
- To determine treatment course?
- To diagnose a patient?
Studies involving gene therapy have additional regulatory requirements and FDA guidances dictating their development. Examples of some key guidances that must be addressed during clinical protocol development include:
- Guidance for Industry- Long Term Follow-up After Administration of Human Gene Therapy Products
- Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus During Product Manufacture and Patient Follow-up
Our CAR T cell protocol templates are written to be in compliance with these and other FDA guidances and to take into account unique operational changes of gene therapy studies:
Extended time from initial patient identification to initiation of treatment (due to manufacturing limitations) resulting in multiple eligibility reviews:
Click to expand image

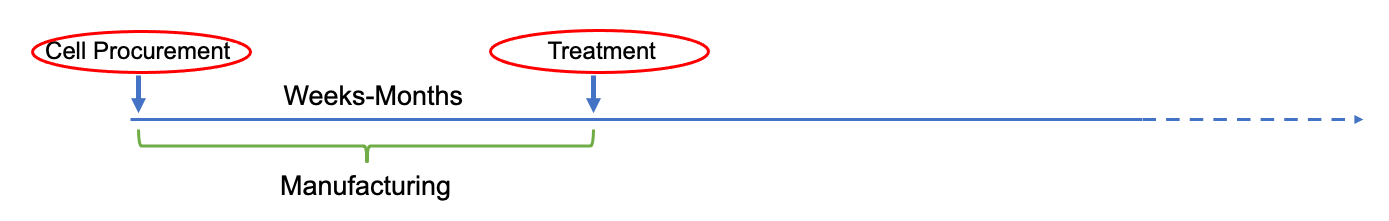
Extended time from initial patient approach to initiation of treatment (due to manufacturing limitations) resulting in stricter eligibility requirement to maintain suitability for treatment:
Click to expand image

Extended time from initial patient approach to initiation of treatment (due to manufacturing limitations) resulting in considerations of larger drop-out rates:
Click to expand image

Extended time from initial patient approach to initiation of treatment (due to manufacturing limitations) resulting in allowances for bridging therapy with special considerations for washout and baseline disease assessments:
Click to expand image

Additional required long-term follow-up for up to 15 years requires consideration about how the follow-up will be accounted for, whether it must be performed at the clinical site and whether different follow-up stages will be treated differently:
Click to expand image

These protocols also require approval from an additional review board, the Institutional Biosafety Committee, when activating the clinical trial.
Under 21 CFR 361.1 you can do basic research using radioactive drugs/biologics in humans without an IND when the drug is administered under the following conditions:
- The research is considered basic science research and is done for the purpose of advancing scientific knowledge:
- Intended to obtain basic information regarding metabolism (including kinetics, distribution, dosimetry, and localization) of a radioactive drug or regarding human physiology, pathophysiology, or biochemistry,
- Not intended for immediate therapeutic, diagnostic or similar purposes (e.g., preventive benefit to the study subject from the research), and
- Not intended to determine the safety and effectiveness of a radioactive drug in humans.
- The study is approved by an FDA-approved Radioactive Drug Research Committee (RDRC)
- The pharmacologic dose of the radioactive drug to be administered is known not to cause any clinical detectable pharmacologic effect in humans.
- The radiation dose to be administered is justified by the quality of the study being undertaken and the importance of the information it seeks to obtain and is within the limits specified in 21 CFR 361.1(b)(3).
UNC has an RDRC to conduct this review. The RCRC approval of a research study is based on assurance that the following requirements are met:
- Appropriate limit on the radiation dose
- Appropriate limit on the pharmacologic dose
- Qualified study investigators
- Medical facility properly licensed to possess and handle radioactive materials
- Appropriate selection and consent of research subjects
- Appropriate quality of radioactive drug administered
- Sound research protocol design
- Reporting of adverse events by the investigator to the RDRC
- Approval by an appropriate Institutional Review Board (IRB)
Other Resources:
- 21 CRF 361.1
- Guidance for Industry and Researchers- The Radioactive Drug Research Committee: Human Research With an Investigational New Drug Application
- Guidance for Industry- Developing Medical Imaging Drug and Biological Products Part 1: Conducting Safety Assessments
- Guidance for Industry- Developing Medical Imaging Drug and Biological Products Part 2: Clinical Indications
- Guidance for Industry- Developing Medical Imaging Drug and Biological Products Part 3: Design, Analysis, and Interpretation of Clinical Studies
- Guidance- PET Drugs—Current Good Manufacturing Practice (CGMP)
It is important to remember that the Federal Food, Drug and Cosmetic Act (FD&C) defines pediatric patients as persons aged 21 or younger at the time of diagnosis or treatment and that the population is further subcategorized:
- Neonates: from birth through the first 28 days of life
- Infants: 29 days to less than 2 years
- Children: 2 years to less than 12 years
- Adolescents: Aged 12 through 21
Based on the drug/biologic that you intend to study, the characteristics of the disease, and the prevalence in the different pediatric subpopulations, you may need to consider separate dose escalations and phase 1 studies in older subjects (particular adults) prior to initiating studies in younger populations.
Studies where you are manufacturing on campus or have an additional institutional Conflict of Interest, such as a patent, require review by the Institutional COI Committee.